


The role of Erastin in ferroptosis
Ferroptosis is a newly discovered way of cell death in recent years, which is mainly characterized by the accumulation of iron-dependent lipid peroxides [1]. There are two main types of ferroptosis inducers: the first type includes Erastin, sulfasalazine, glutamate, etc., which can act through system xc-; the second type includes RSL3, DP17, etc., which can directly inhibit glutathione Peptide peroxidase (GPX) activity plays a role [2]. Among them, Erastin and other ferroptosis inducers usually mediate a single pathway, it can mediate a variety of molecules, and the effect is efficient, rapid and durable [3]. This article reviews the action pathway and anti-tumor characteristics of Erastin.
1. The discovery of Erastin
In 2003, Dolma et al. [4] discovered a small molecule compound that can selectively kill engineered tumor cells expressing ST and RAS, and named it Erastin. However, unlike the apoptosis of ST and RAS-expressing cells induced by camptothecin, the classic features of apoptosis such as release of mitochondrial cytochrome c, activation of Caspase-3, and DNA fragmentation were not found in cells that were induced to die by Erastin. And this mode of death cannot be inhibited by apoptosis inhibitors [4,5,6]. Therefore, what Erastin induces is a novel, non-apoptotic form of cell death [5]. According to its characteristics, Dixon et al. [1] named Erastin-induced cell death as ferroptosis in 2012.
2. Characteristics and related pathways of ferroptosis
2.1 The concept and characteristics of iron death
Ferroptosis is a form of cell death manifested by iron-dependent accumulation of intracellular lipid reactive oxygen species (L-ROS). There are significant differences between ferroptosis and other cell death methods (such as apoptosis, necrosis, autophagy, etc.) in many aspects. Morphologically, ferroptotic cells exhibit characteristic mitochondrial shrinkage and increased mitochondrial membrane density, whereas other forms of cell death typical of ferroptosis are absent [1,7]. In terms of biochemical metabolism, redox homeostasis in ferroptotic cells is broken, antioxidant capacity is reduced, and intracellular reactive oxygen species are increased. At the same time, this death process can be inhibited by antioxidants and iron chelators [1]. Although there are multiple upstream pathways that induce ferroptosis, they will eventually lead to the imbalance of intracellular L-ROS generation and degradation, resulting in increased L-ROS and ferroptosis [1,8].
2.2 Induction of ferroptosis by inhibiting the cystine-glutamate transport receptor system xc-
system xc- is an antiporter composed of SLC7A11 and SLC3A2 subunits, which can transfer glutamate out of cells and cystine into cells [9]. Cystine transferred into cells is involved in the synthesis of glutathione (GSH). GPX plays a major role in maintaining redox homeostasis in cells, and GSH is a cofactor for its activation [10]. A variety of ferroptosis inducers can inhibit the absorption of cystine by inhibiting system xc-, resulting in decreased GPX activity, decreased cellular antioxidant capacity, increased intracellular L-ROS, and ferroptosis [1].
2.3 Participation of p53 in ferroptosis and other pathways mediated
p53 is a classic tumor suppressor gene that also plays an important role in inducing ferroptosis in cancer cells. After the activation of the p53 gene, it inhibits the expression of SLC7A11 and thus inhibits the activity of system xc-, which reduces the antioxidant capacity of cells, thereby triggering ferroptosis [11].
GPX4 is a member of the GPX family and plays an important role in maintaining intracellular redox homeostasis [8]. Inducers such as RSL3 can directly inhibit the activity of GPX4 and cause ferroptosis [1]. Voltage-dependent anion channels (VDACs) are exchange channels on the mitochondrial outer membrane, which can be induced by drugs to change permeability, causing mitochondrial metabolic disorders, reactive oxygen species production, and oxidative death [12].
3. Erastin triggers ferroptosis by altering VDAC permeability
3.1 Structure and regulatory function of VDAC
VDAC is a channel in the mitochondrial outer membrane that controls the flow of metabolites across the membrane [13]. Following breakdown of respiratory substrates, mitochondrial membrane potential (ΔΨ) is maintained and used to generate adenosine triphosphate (ATP). In the open state, VDAC allows the entry of respiratory substrates, adenosine diphosphate (ADP), and phosphate (Pi) into the mitochondria, and in the closed state, closes the mitochondria [14]. The dynamic "open-close" of VDAC will have a major impact on mitochondrial metabolism and cellular bioenergetics.
3.2 Inhibitory effect of tubulin on VDAC
Tubulin is a globular protein that dynamically regulates mitochondrial metabolism by blocking VDAC [12]. After VDAC is blocked by tubulin and shuts down, it restricts the influx of metabolites into mitochondria and ATP production, and subsequently mitochondrial metabolism is inhibited (ΔΨ decreases) and maintains a lower ATP/ADP ratio, which favors aerobic glycolysis in cancer cells, leading to Warburg effect [15].
3.3 Warburg effect
Cells obtain energy mainly through oxidative phosphorylation under aerobic conditions, and mainly through glycolysis under hypoxia [16]. However, malignant tumor cells still use glycolysis as a source of energy when oxygen levels are sufficient, which is called "Warburg metabolism" [17]. Warburg metabolism is a metabolic signature that favors cancer cell proliferation [18]. In cancer cells, free tubulin blocks VDAC, resulting in inhibition of mitochondrial function. Therefore, decreased mitochondrial metabolism, increased glycolysis, and decreased ATP/ADP ratio are the main features of Warburg metabolism.
3.4 Erastin antagonizes tubulin to cause anti-Warburg effect and double-hit effect
Erastin can specifically inhibit the function of tubulin on VDAC, prevent the blockage of VDAC by free cytoplasmic tubulin, and make VDAC open [5]. VDAC opening causes increased mitochondrial metabolism (ie, increased ΔΨ), decreased glycolysis, and increased reactive oxygen species production. This triggers two effects: increased production of reactive oxygen species leads to oxidative stress (first hit); with reduced glycolysis, increased oxidative phosphorylation and ATP synthesis leads to a reversal of the Warburg effect (second hit) . The anti-Warburg effect can damage or reduce cancer cell proliferation [18]. One of the advantages of Erastin as a VDAC-tubulin antagonist is the specific killing of cancer cells because in non-proliferating cells VDAC is structurally open and not regulated by tubulin [19]. Therefore, this represents a new pharmacological target, and Erastin may become a novel anticancer drug that acts by regulating metabolism (Figure 1).
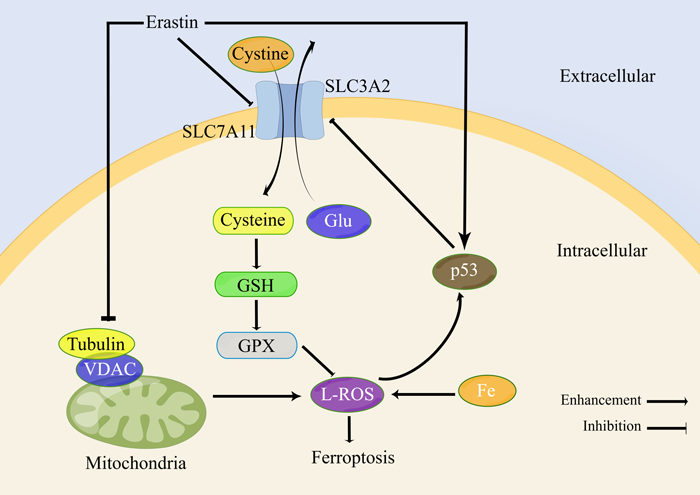
Figure 1.Erastin-induced ferroptosis-related pathways
Note: SLC3A2 and SLC7A11 are cystine-glutamate transporters; GSH is glutathione; VDAC is voltage-dependent anion channel; GPX is glutathione peroxidase; L-ROS is lipid reactive oxygen species
4.Erastin acts on system xc- and activates p53 to induce ferroptosis
Studies have confirmed that Erastin reduces cellular uptake of cystine by directly inhibiting system xc-activity [1,19]. As a raw material for the synthesis of GSH, the reduction of cystine uptake will inevitably lead to the reduction of intracellular GSH synthesis. It was found that GSH in cells was significantly depleted after Erastin treatment [2,20]. GSH is an essential cofactor for GPX4 to inhibit L-ROS production, so the inhibition of system xc- by Erastin indirectly leads to the reduction of GPX4 synthesis and the reduction of cellular antioxidant capacity [1]. Experiments found that the activity of GPX4 was decreased in a variety of cancer cells treated with Erastin, and the cell death caused by Erastin inhibition of system xc- or GPX4 inactivation involved iron-dependent accumulation of L-ROS and consumption of polyunsaturated fatty acids. [1,2,20]. Therefore, Erastin prevents the entry of extracellular cystine into cells by inhibiting system xc- and reduces the level of intracellular GSH, which is an essential substrate for GPX4 to exert its antioxidant effect, thus reducing GPX4 activity and disrupting cellular redox homeostasis , L-ROS accumulation, causing ferroptosis.
Activation of the p53 gene inhibits system xc-activity and triggers ferroptosis. Studies have shown that Erastin can also enhance ferroptosis by activating p53. After Erastin treatment of lung cancer A549 cells, the transcription of p53 was significantly up-regulated, and the level of reactive oxygen species was significantly increased. Thus, Erastin exposure generates reactive oxygen species, which activate p53, which subsequently acts on downstream pathways. More importantly, Erastin caused an increase in reactive oxygen species, followed by p53 activation. As a feedback loop, the activated p53 in turn caused an increase in reactive oxygen species, aggravating the cytotoxic and cytostatic effects of Erastin on A549 cells [21] (Fig. 1).
5.Erastin pharmacodynamics and safety evaluation
Yang et al[2] established a xenograft mouse model and evaluated the efficacy of the Erastin analogue PE. The tumor-bearing mice were injected with PE subcutaneously and through the tail vein at 40 mg/kg each time and 30 mg/kg each time, respectively. It was observed that the tumor growth in the PE treatment group was significantly delayed compared with the blank control group. In addition, 60 mg/kg of PE was injected into the tail vein to evaluate its systemic adverse reactions and pharmacodynamics. At this dose, no obvious adverse reactions were observed, no acute lethal injury or significant body weight loss was found, which proved that the dose was acceptable. In the in vivo experiments that have been carried out, tumor growth was inhibited after administration of Erastin. Pharmacodynamic and toxicological studies have shown that the drug is tolerated at potentially therapeutic levels based on body surface area ratios [22]. Given that most of the current research on Erastin comes from cell experiments, more in vivo experiments need to be carried out.
6. Feasibility of clinical application of Erastin
6.1 Application in chemotherapy
The continuous and extensive use of chemotherapeutic drugs will lead to tumor resistance [23,24], and inhibition of tumor cell apoptosis or reduced susceptibility is an important mechanism of drug resistance [25]. So, could drug resistance be overcome by other non-apoptotic means? As mentioned above, Erastin causes cancer cell death to be non-apoptotic iron-dependent death. In addition to inducing iron death by itself, Erastin combined with chemotherapeutic drugs can enhance the chemosensitivity of cancer cells [26]. Erastin can respectively enhance the sensitivity of lung cancer cells to cisplatin [27], rhabdomyosarcoma cells to doxorubicin and actinomycin D [28], and glioblastoma cells to temozolomide [29]. In addition, Erastin can also eliminate the drug resistance of multiple chemotherapy-resistant cells [30], which provides more feasibility for Erastin as an anticancer drug.
6.2 Application in radiotherapy
Radiation therapy is the second largest treatment for malignant tumors after surgery. How to increase the sensitivity of tumors to radiation to improve the efficacy of radiation therapy is an urgent problem to be solved.
Radiosensitizers can increase the radiosensitivity of tumor cells [31,32]. In addition to ferroptosis-inducing effects, Erastin can also increase the sensitivity of cancer cells to radiation. Erastin can increase the sensitivity of breast cancer cells to γ-rays by inhibiting system xc-, prolonging the damage of radiation to DNA [33]. More advantageously, most normal cells do not express SLC7A11 [33], so Erastin inhibition of system xc- can specifically enhance the radiosensitivity of tumor cells and protect normal cells from radiation damage, which has great clinical application value.
7. Summary and Outlook
Erastin is a small molecule compound that can specifically kill tumor cells in a rapid and irreversible process. As a ferroptosis inducer, Erastin can mediate multiple pathways, unlike other ferroptosis inducers that usually mediate a single pathway, inhibiting system xc-cystine-glutamate transport, acting on VDAC to relieve microtubules The inhibitory effect of the protein on VDAC may indirectly inhibit system xc- by activating the p53 gene, resulting in the occurrence of ferroptosis. Erastin acts more efficiently, rapidly, and lasts longer than other ferroptosis inducers, and requires only very low concentrations.
More importantly, Erastin can enhance the chemosensitivity of various cancer cells and enhance the radiosensitivity of cancer cells, so it can be used as a new type of chemotherapeutic drugs or radiosensitizers in clinic. However, given the insufficient number of in vitro and in vivo studies on Erastin, more experiments need to be performed.








Comments